

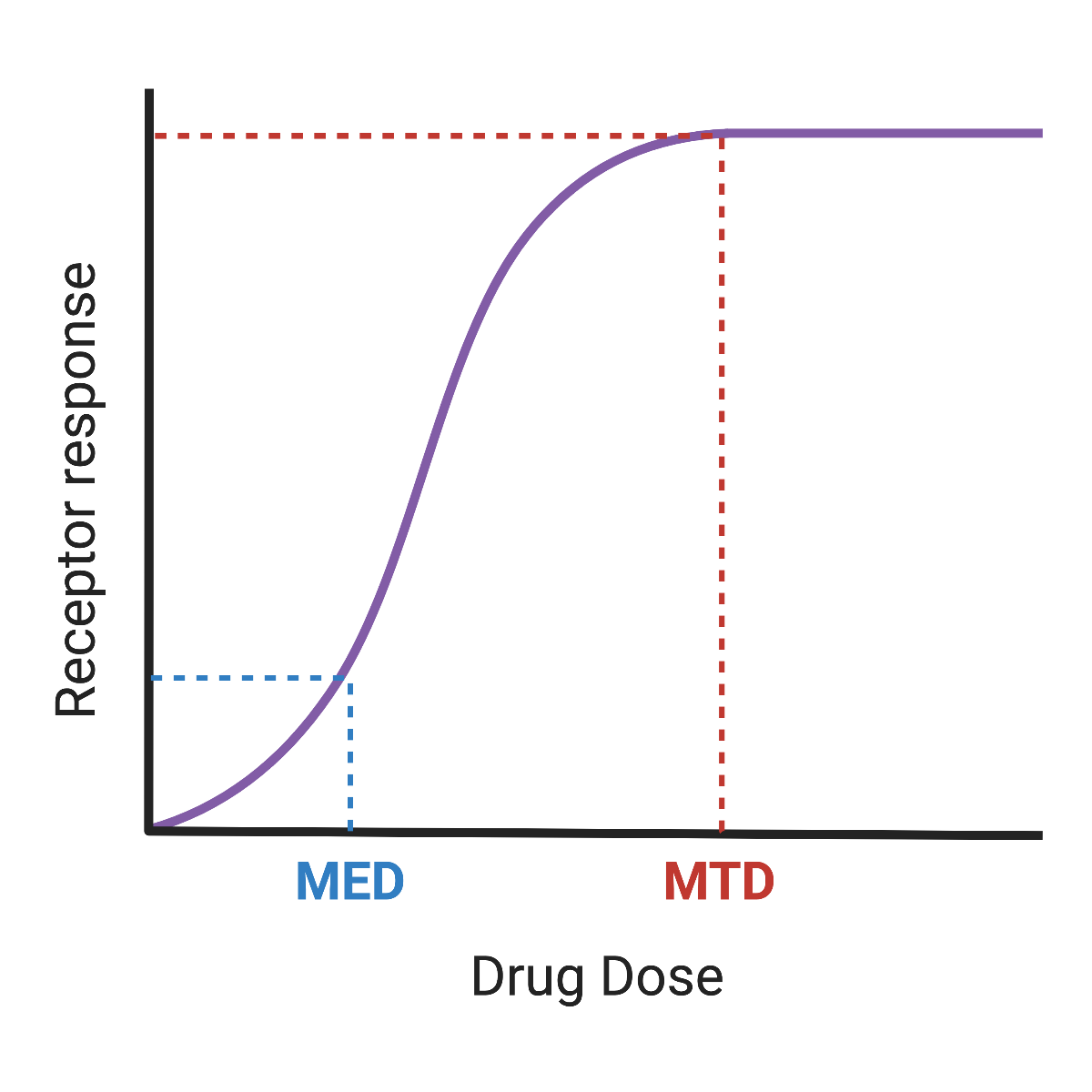
Dosage is one of the key components of an effective application of therapeutic drugs in patients. Too low a dosage may lead to a poor response in the patient. However, unnecessarily high dosage is wasteful and could be toxic to the patient. Today’s blog will focus on how drug developers decide on the proper dosage. Specifically, we’ll talk about Maximum Tolerated Dose (MTD) vs Minimum Effective Dose (MED).
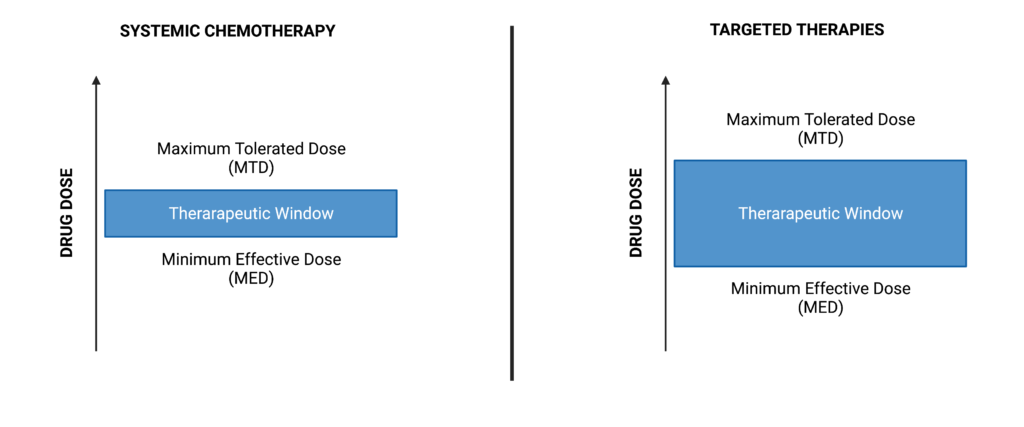
In the US, the Federal Drug Administration (FDA) used the concept of the MTD to guide dosage recommendations in oncology clinical trials. MTD came into fruition alongside broad-range cytotoxic chemotherapies. The goal was to provide the highest possible dosage to patients to maximize efficacy before reaching a “dose-limiting toxicity” (DLT) at which point the safety of the patient was compromised. Phase I trials today often use a “3+3 design” in which sets of 3 patients are given successively higher doses until two of the 3 patients exhibit DLT; the next lowest dosage becomes the MTD. Future trials often continue with this dosage as the default, despite the fact the dose may be high enough to still inflict significant discomfort and reduced quality of life.
Over the past decade, momentum has been building for a paradigm shift in dose determination. Modern cancer therapies, such as monoclonal antibodies and CAR T-cells, are highly targeted and may not benefit from the “more is better” approach used in chemotherapies. Discussions from organizations such as the American Society of Cancer Research (ACSR) and American Association for Cancer Research (AACR) have pressed for dose determination based on efficacy, not tolerance, which has culminated in the FDA’s recent “Project Optimus” campaign. Under this program, pharmaceutical companies are being encouraged to incorporate Minimum Effective Dose (MED) analyses into their studies. When evaluating MTD vs MED, switching to an MED will lead to improved quality of life for patients.
Switching to an MED framework will require an improved ability to rapidly and accurately assess whether a dose is effective. Measuring changes in tumor size are one way to assess effectiveness but requires longer wait times. An alternative method of assessing MED that goes hand in hand with targeted therapies is to directly quantify activation states of proteins in the targeted pathway. This method requires in-depth knowledge of the target and pathway involved. Techniques such as Western blots and IHC can be used toward this end but are often laborious and can detect a limited number of analytes per experiment. In contrast, reverse phase protein arrays (RPPA) allow for the detection of dozens of proteins and their activation states rapidly, which is key when trying to see how an entire pathway is responding to a treatment dosage.
Currently, we offer the Theralink Assay for Breast Cancer, a CLIA-approved RPPA assay that can be used in the clinic to guide physicians in selecting the best therapies. We are also capable of fully customizing an RPPA experiment to analyze a wide number of pathways, with 700 targets currently validated and more being added all the time. Given the FDA’s future vision of selecting dosage by efficacy and not just tolerance, we look forward to playing a pivotal role in transforming the way therapies are approved.